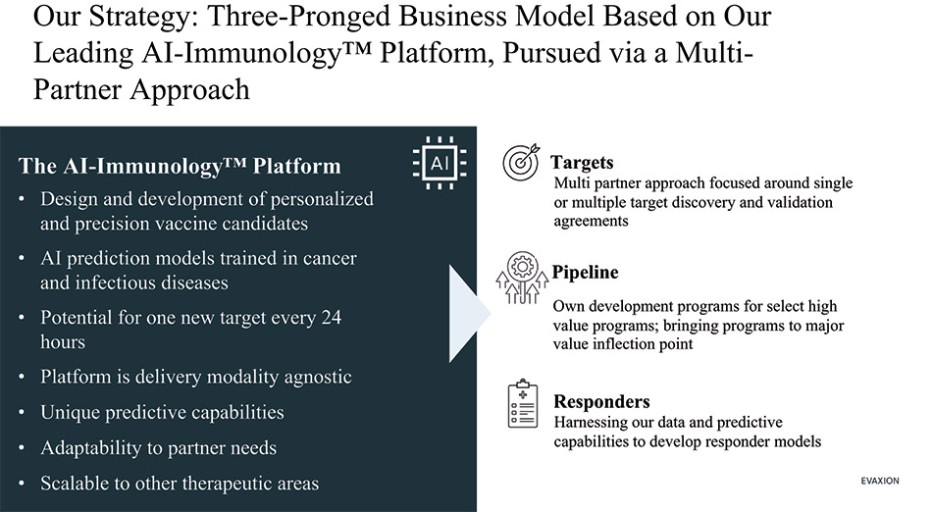
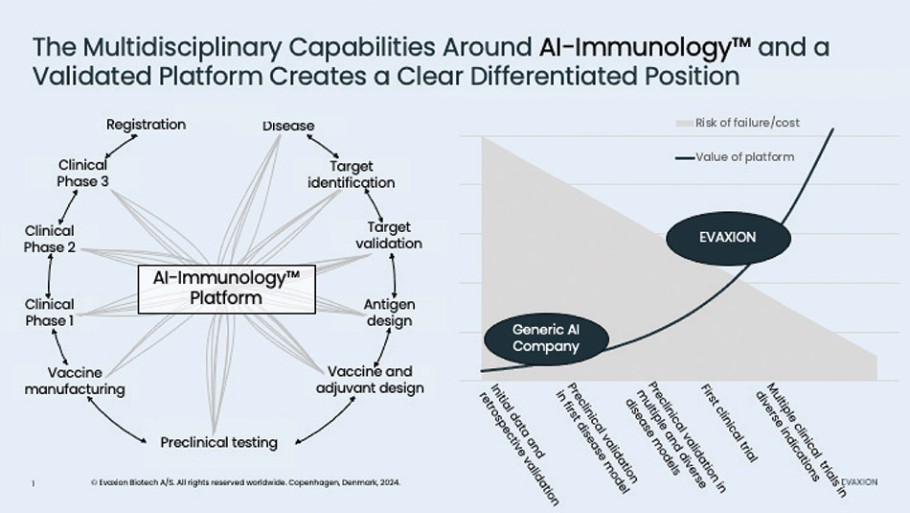
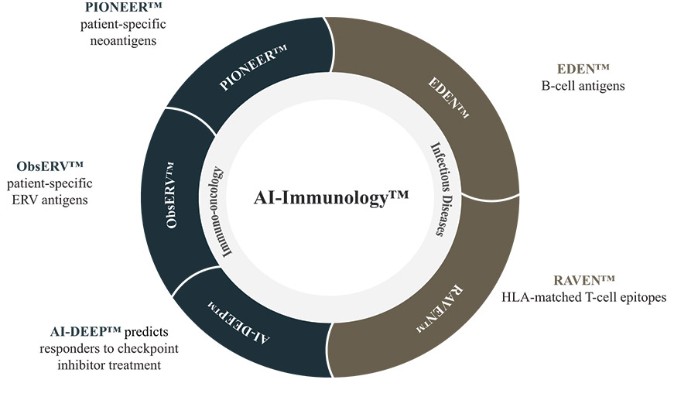
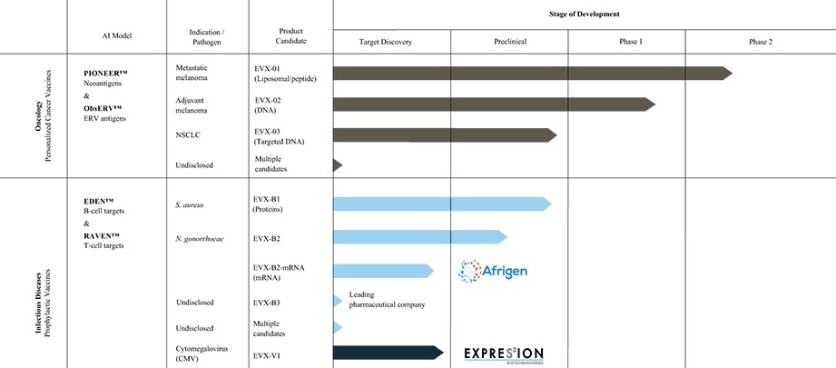
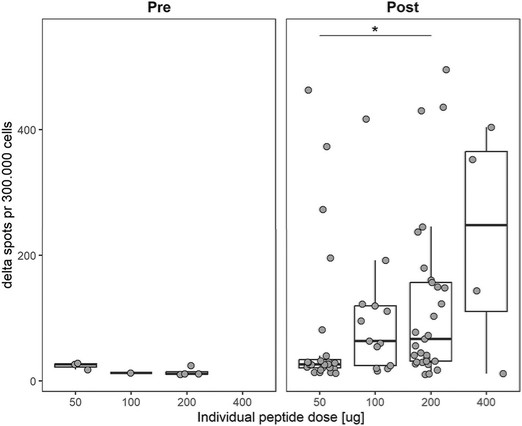
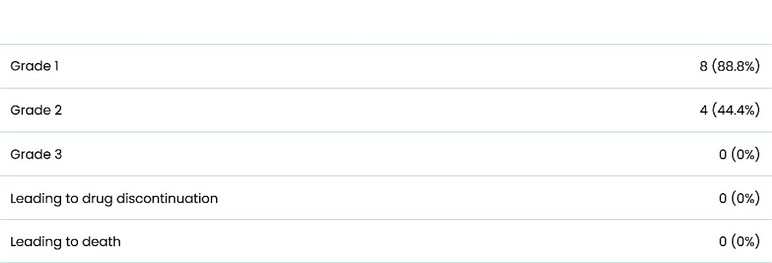
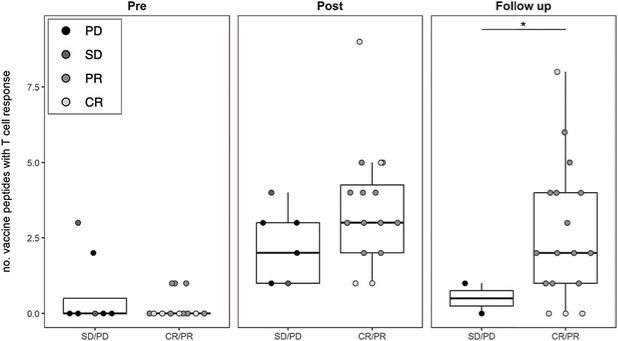
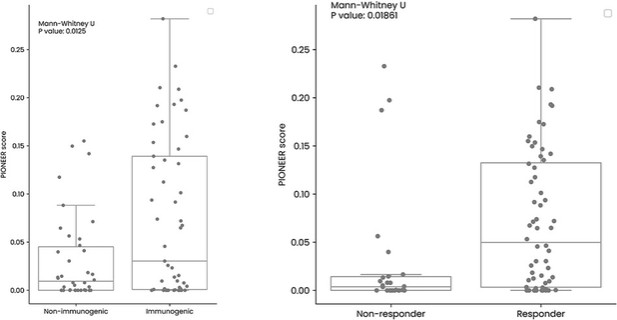
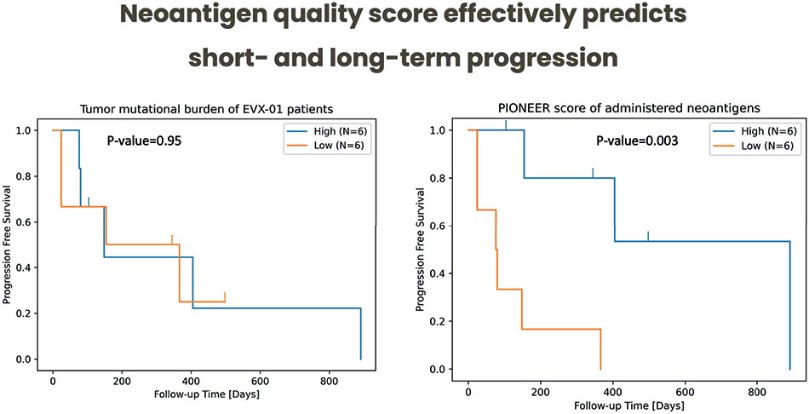
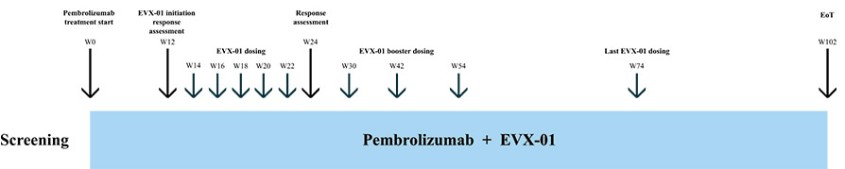
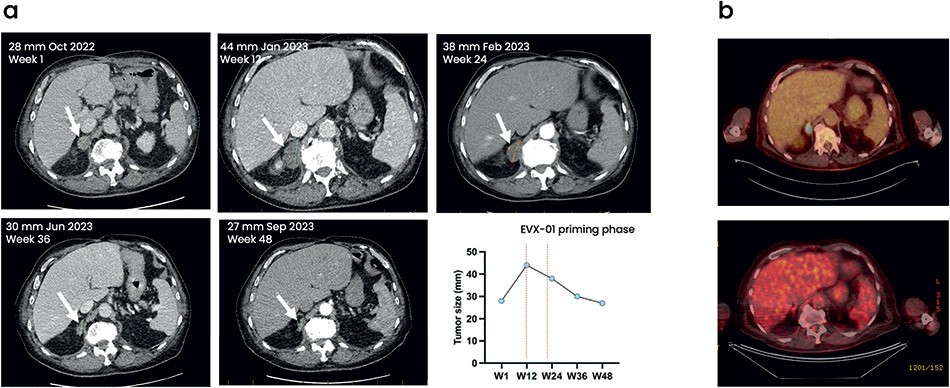
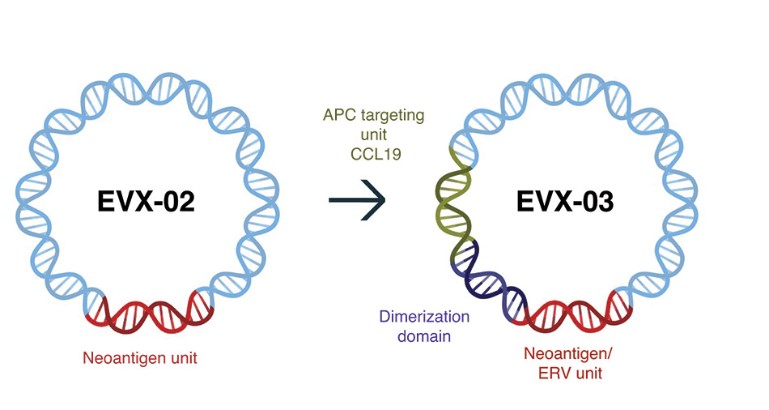
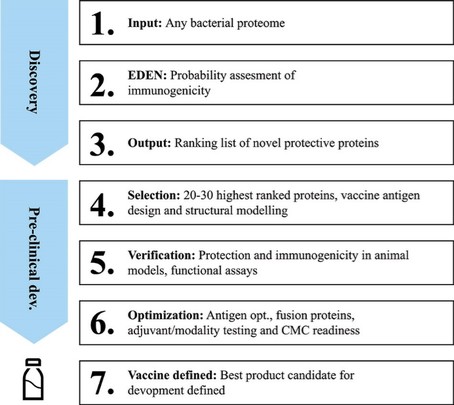
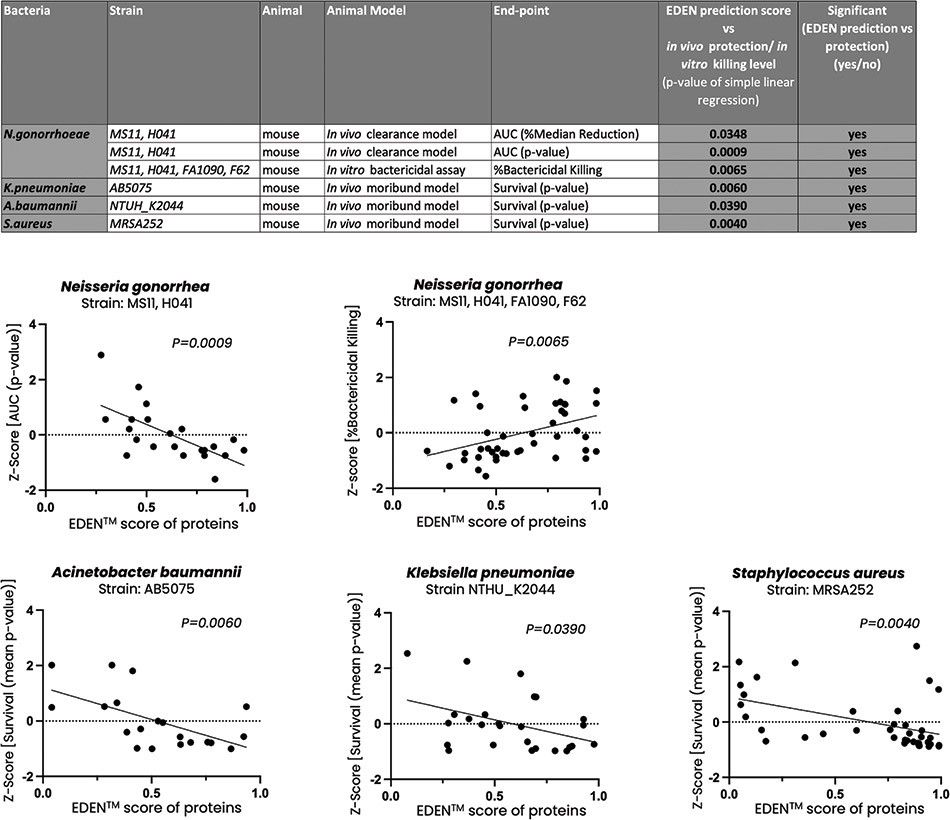
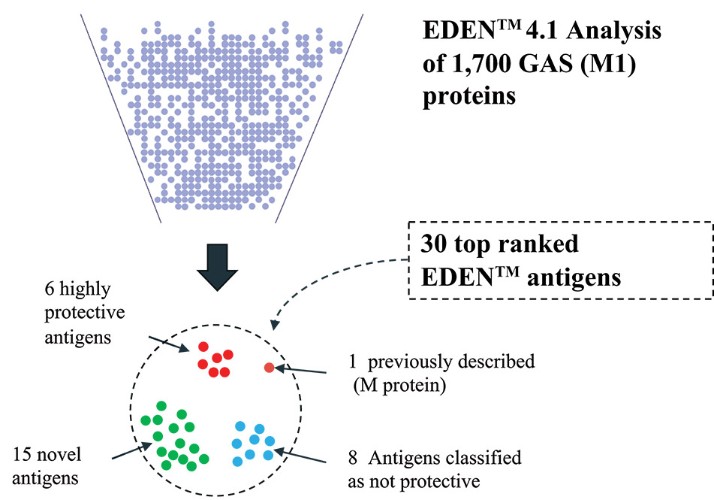
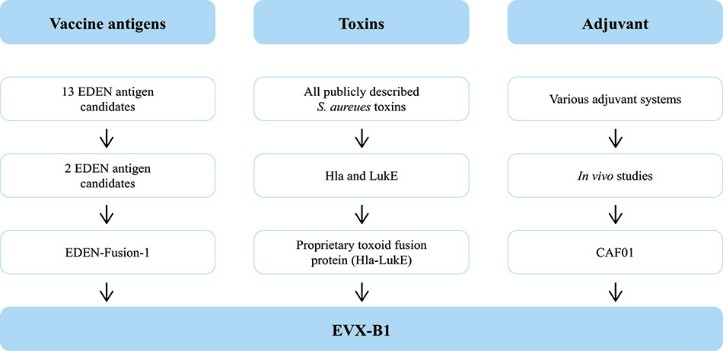
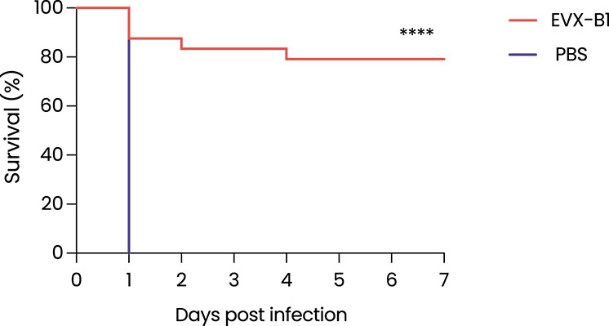
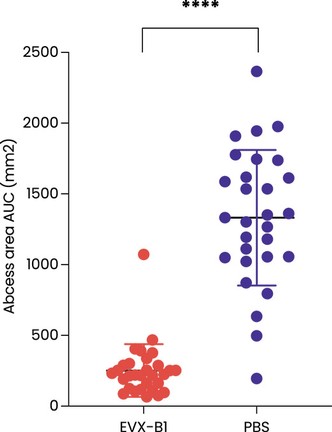
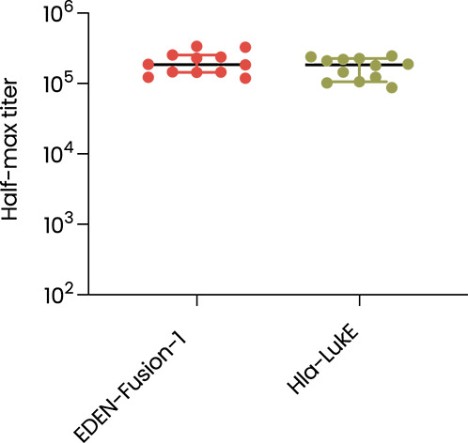
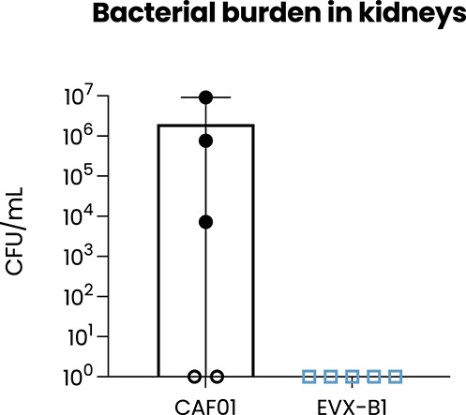
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
PRELIMINARY PROSPECTUS SUBJECT TO COMPLETION DATED , 2024
UP TO 1,326,260 AMERICAN DEPOSITARY SHARES REPRESENTING
13,262,600 ORDINARY SHARES
AND UP TO 1,326,260 PRE-FUNDED WARRANTS TO PURCHASE UP TO
1,326,260 AMERICAN DEPOSITARY SHARES
AND UP TO 1,326,260 SERIES A WARRANTS TO PURCHASE UP TO
1,326,260 AMERICAN DEPOSITARY SHARES
AND PLACEMENT AGENT WARRANTS TO PURCHASE UP TO 66,313 AMERICAN
DEPOSITARY SHARES
(and 1,326,260 American Depositary Shares representing 13,262,600 ordinary shares underlying the Pre-Funded Warrants and 1,326,260 American Depositary Shares representing 13,262,600 ordinary shares underlying the Series A Warrants and 66,313 American Depositary Shares representing 663,130 ordinary shares underlying the Placement Agent Warrants)

Evaxion Biotech A/S
We are offering 1,326,260 American depositary shares, or ADSs representing 13,262,600 ordinary shares, DKK 1 nominal value per share, together with Series A warrants to purchase up to 1,326,260 ADSs representing 13,262,600 ordinary shares (the “Series A Warrants”, or the “Warrants”). The ADSs and Warrants will be sold in a fixed combination, with each ADS accompanied by one Series A Warrant to purchase one ADS. The ADSs and Warrants are immediately separable and will be issued separately in this offering, but must be purchased together in this offering. The assumed public offering price for each ADS and accompanying Warrant is $7.540, which is based upon the closing price of our ADSs on The Nasdaq Capital Market on January 10, 2024 (after giving effect to the ADS ratio change as further described below). The Series A Warrants will have an exercise price per share of $ and will be immediately exercisable for a term of five (5) years from the date of issuance. The recent market price used throughout this prospectus may not be indicative of the final offering price. The final public offering price will be determined through negotiation between us and investors based upon a number of factors, including our history and our prospects, the industry in which we operate, our past and present operating results, the previous experience of our executive officers and the general condition of the securities markets at the time of this offering.
We are also offering to certain purchasers whose purchase of ADSs in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding ADSs immediately following the consummation of this offering, the opportunity to purchase, if any such purchaser so chooses, pre-funded warrants, in lieu of ADSs that would otherwise result in such purchaser’s beneficial ownership exceeding 4.99% (or, at the election of the purchaser, 9.99%) of our ADSs. The public offering price of each pre-funded warrant will be equal to the price at which an ADS is sold to the public in this offering, minus $0.1479 or the USD equivalent to DKK 1 at the time of conversion, and the exercise price of each pre-funded warrant will be $0.1479 per ADS or the USD equivalent to DKK 1 at the time of conversion, provided that such exercise price may be paid at the time of issuance and held in escrow until the time of conversion. The pre-funded warrants will be immediately exercisable and may be exercised at any time until all of the pre-funded warrants are exercised in full. For each pre-funded warrant we sell, the number of ADSs we are offering will be decreased on a one-for-one basis. The ADSs and pre-funded warrants can only be purchased together in this offering but will be issued separately and will be immediately separable upon issuance. This prospectus also relates to the ADSs issuable upon exercise of the Series A Warrants and any pre-funded warrants sold in this offering.
There is no established public trading market for the Warrants and pre-funded warrants, and we do not expect a market to develop. We do not intend to apply for listing of the Warrants and pre-funded warrants on any securities exchange or other nationally recognized trading system. Without an active trading market, the liquidity of the warrants will be limited.
This offering will terminate on [*], 2024, unless we decide to terminate the offering (which we may do at any time in our discretion) prior to that date. We will have one closing for all the securities purchased in this offering.
Our ADSs are listed on the Nasdaq Capital Market, or Nasdaq, under the symbol “EVAX”. On January 10, 2024, the closing trading price for our ADSs, as reported on Nasdaq, was $7.540 per ADS (after giving effect to the ADS ratio change as further described below).